用 AI 识别基因,从向量化 DNA 序列开始
DNA 序列在分子生物学和医药研究中有着广泛的应用,比如基因溯源、物种鉴定、疾病诊断等。如果结合正在兴起的基因大数据,采取大量的样本,那么通常实验结果更具说服力,也能够更有效地投入现实应用。
同时如同其他行业一样,人工智能的介入正在受到广泛的关注,承载着业界对更智能高效的研究方法的期待。然而传统的核酸序列比对方法有着诸多限制,并不适用于大规模的数据 [1] ,这使现实应用不得不在成本和准确率中做出取舍。为缓解核酸序列数据特性的掣肘,向量化是面对大量 DNA 序列时的一个更优选择。
Milvus 作为一款开源的、对海量数据友好的向量数据库,能够高效地存储和检索核酸序列的嵌入。在提高效率的同时,Milvus 也能够帮助降低项目研究或系统搭建的成本。由 Milvus 搭建的 DNA 序列分类系统,不仅毫秒之间能够识别基因的类别,还比机器学习领域里常见的分类器们更加精准。
数据处理
基因是带有遗传信息的 DNA 序列片段,由数个碱基【A, C, G, T】排列组合而成。每个生物都有不同的基因组,比如人类基因组中含有3万个左右基因,约30亿个 DNA 碱基对,每个碱基对有2个对应的碱基。
针对不同的需求和目的,DNA 序列可以被各种分类,支持着多样的学术研究和现实应用。原始的 DNA 序列数据通常长短不一,常存在长序列。为了减少处理数据的成本,业内通常使用 k-mer [2] 预处理序列,同时能够使 DNA 序列更接近普通文本的词句结构。而向量化数据则能够进一步提高计算速度,并适用于大数据分析与机器学习。
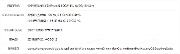
k-mer
一种常见的 DNA 序列预处理方式是 k-mer,从原始序列第一个碱基开始,以一个碱基为单位每次向后一位,每次取一个长度为k的短序列。经过 k-mer 之后,一条长度为 s 的长序列就被转换成了(s-k+1)个短序列。通过调节k的值,可以提高模型的准确性。转换后的短序列可以更好地进行数据读取、特征提取、向量化。
向量化
向量化 DNA 序列的过程其实是将其当作普通文本,一条被 kmer 拆分后的序列就像是一个句子,拆成的单个短序列是一个单词,碱基则对应字符。因此常见的 NLP 模型都可以被用来处理 DNA 序列数据,进行模型训练、特征提取、序列编码。
每个模型都有自己的特点和限制,实际操作中可以根据数据的特点进行选择和比较。以词袋模型为例,CountVectorizer 是一种比较轻便的特征提取方法,对序列长度没有限制,但相似度区分不明显。
Milvus示例
Milvus 使用简化的非结构化数据管理,能够将目标对象在万亿条向量数据中根据近似最近邻搜索(ANN)算法进行比对,以平均延迟以毫秒计的速度召回相似结果。它对海量非结构化数据的友好与高效的相似搜索,毫无疑问能够轻松地管理大量 DNA 序列数据,从而促进生物或基因学的研究与应用。
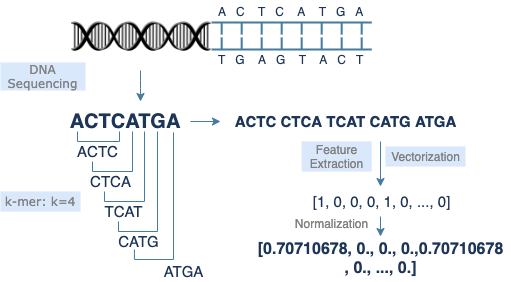
一个简单的演示案例展现了如何使用Milvus搭建 DNA 序列的分类系统,实验数据[3]包含了三个物种的7种基因序列。在插入 Milvus 之前,该示例首先将所有的 DNA 序序列进行了 k-mer 处理,然后训练了词袋模型用以特征提取与向量化。该结合 Milvus 与 Mysql 的分类模型结构如下图所示,包括了插入和搜索两个流程。
(https://github.com/milvus-io/bootcamp/tree/master/solutions/dna_sequence_classification)
得到每条 DNA 序列对应的向量之后,将其插入 Milvus 中事先建好的 Milvus 集合中(可指定分集),以供比对与搜索。向量之间的距离反映了序列之间的相似度,从而实现基因序列的分类和种类之间的相似度比较。

基因序列分类
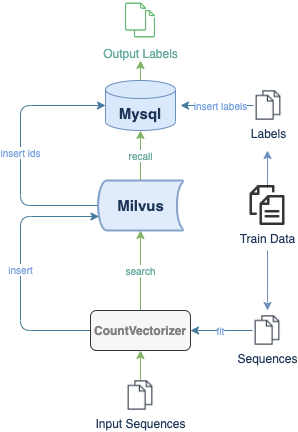
输入未知类别的 DNA 序列,在 Milvus 数据库中搜索与其相似的序列便可以对搜索对象进行基因分类,以此了解其可能的功能。比如一段序列被分类为GPCRs(G蛋白偶联受体),则表示该序列或许关系着样本采取对象的各种生理功能[4]。示例中的模型对未插入集合的人类基因序列进行搜索,成功根据搜索结果返回了正确的基因类别,证明了 Milvus 的向量相似性搜索能够分类基因序列。
物种相似程度
通过比较不同物种之间基因序列的平均相似度,也可以看出哪些物种之间基因更为相似。示例中计算了黑猩猩与人类、狗与人类的基因序列的平均内积距离(Average Inner Product Distance: 0.97 > 0.70),验证了黑猩猩比狗在基因上更接近人类。由此可见,Milvus 能够通过计算基因序列之间的向量距离来支持科学研究。
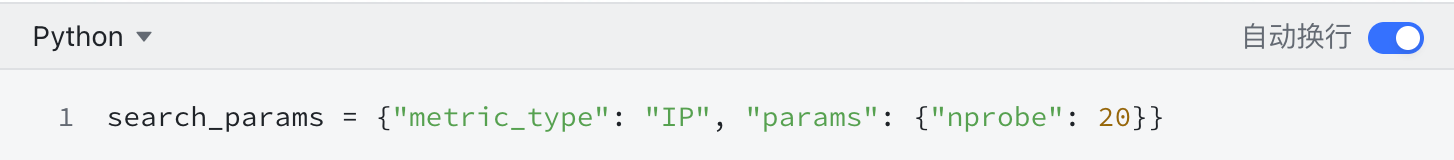
实验表现
对示例采用的人类基因序列样本数据(共 3629 条)进行 k-mer 处理后,随机取20% 作为测试数据。使用剩下的 80% 作为训练数据,进行特征提取,训练不同的分类模型或搭建 Milvus + Mysql 搜索系统。实验采用了五种分类器分别对测试序列进行了分类,通过计算并比较准确率(正确分类的基因序列个数 / 测试序列的总个数),Milvus + Mysql 搭建的分类模型的准确率(90.77%)超过了所有的常用分类器。


应用拓展
随着基因大数据的发展和完善,向量化后的 DNA 序列数据能够更好地参与科学研究与实践应用。如果能够结合生物学的专业知识,便可以更合理地向量化 DNA 序列、计算距离、解读结果。
融入生物学并采用大量的样本能够很大程度地改进基因序列的向量数据库,Milvus 便能够更好地发挥所长。根据现实需求,Milvus 相似性搜索和距离计算有很大的潜力被投入各种应用。
-
未知序列研究:研究表明向量化序列能够压缩数据,根据已知基因序列研究未知序列的结构、功能、进化关系。[5] 当拥有足够的序列数据进行研究时,实验结果会更加可靠有效,但数据的存储和处理会成为一个问题。将其向量化后使用 Milvus 能够在节约数据处理成本的同时,保持甚至提高模型准确率。
-
适配硬件:受到传统的生物分子序列比对算法限制,基因序列相似性搜索无法受益于硬件(CPU/GPU)的发展[6][7]。Milvus 的加入可以从距离算法上解决这一问题,更好地根据数据规模适配硬件,从而显著提高搜索效率。
-
鉴定或溯源病毒:现实中鉴定新冠病毒的起源时,科学家通过比较毒株核苷酸序列推测出新冠病毒或起源于蝙蝠,同时发现其比起 MERS 更接近 SARS 病毒。[8] 该实验采用了 5 例病患的数据,如果在此基础上使用更大的样本进行验证或者研究,结论能够更具说服力,或发现更多的模式。
-
疾病诊断:临床上一般是对比检查对象与健康人的基因序列,找出可引起疾病的变异基因。[9] 在疾病对应的基因位置得到序列,根据健康与否、严重程度或疾病类型将大量的样本数据分类。将这些数据通过合适的算法提取特征并向量化后插入 Milvus,计算向量距离并转换成患病概率,就可以实现基于基因序列的人工智能疾病诊断系统。除了能够协助疾病诊断外,该应用也能够帮助推进靶向治疗的发展[10]。
参考资料
https://www.frontiersin.org/articles/10.3389/fbioe.2020.01032/full#h5
https://en.wikipedia.org/wiki/K-mer#:~:text=Usually%2C%20the%20term%20k%2Dmer,total%20possible%20k%2Dmers%2C%20where
https://www.kaggle.com/nageshsingh/dna-sequence-dataset
https://baike.baidu.com/item/G%E8%9B%8B%E7%99%BD%E5%81%B6%E8%81%94%E5%8F%97%E4%BD%93/9495289#4
https://iopscience.iop.org/article/10.1088/1742-6596/1453/1/012071/pdf
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7884812/
https://mjeer.journals.ekb.eg/article_146090.html
http://rs.yiigle.com/yufabiao/1180150.htm
http://www.xinhuanet.com/science/2018-04/16/c_137114251.htm
https://www.frontiersin.org/articles/10.3389/fgene.2021.680117/full
✏️ 作者:顾梦佳,Zilliz 数据工程师,麦吉尔大学信息学硕士。在 Zilliz 的主要工作是参与开源项目 Milvus 的社区运营与维护,为用户提供升级推荐系统、搭建基因序列分类模型等不同场景下的解决方案。野草般顽强,解决问题时百折不挠、遇强愈强。
|








- 上一条: 带你了解NB-IoT标准演进 2021-08-19
- 下一条: 微服务体系中的分层设计和领域划分! 2021-08-19
- 用100行代码提升10倍的性能 2021-07-05
- 用AI给向量检索加buff!Milvus亮相数据库顶会VLDB 2021-08-27
- 用 AI 给向量检索加 buff,Milvus 亮相数据库顶会 VLDB 2021-08-27
- AI 框架部署方案之模型量化的损失分析 2021-11-04
- Paddle.js & PaddleClas 实战 ——『寻物大作战』AI 小游戏 2021-11-29